2023年1月6日,新年伊始,欧盟委员会正式通过了MDR过渡期延长法案,发布了《Brussels, 6.1.2023 2023/0005 (COD), amending Regulations (EU) 2017/745 and (EU) 2017/746 as regards the transitional provisions for certain medical devices and in vitro diagnostic medical devices》,该提案的通过给予了制造商更多时间来认证医疗器械,以降低医疗资源短缺的风险。
一、法规背景
欧盟医疗器械(EU)2017/745(MDR)和体外诊断(EU)2017/746(IVDR)新法规分别于2021年5月26日和2022年5月26日正式生效。新法规为医疗器械和体外诊断建立了更为严格的监管体系。受限于产品种类繁多、公告机构容量不足,制造商准备不足和新冠肺炎疫情的影响等,很多MDD下的产品还没有成功切换为MDR。如下图所示,公告机构颁发的MDR证书数量远小于制造商的申请数量,预计到 2024年5月颁发的MDR 证书数量可能仅7000 份左右,而目前市场上共计17095个证书将在 2024 年5月份前到期。因此,实行过渡期延长法案迫在眉睫。
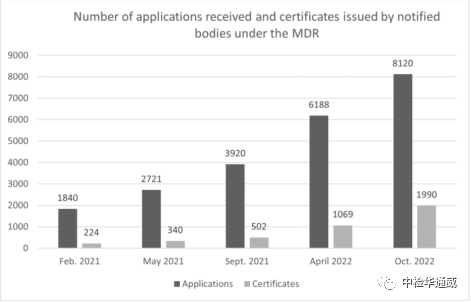
MDR实施后收到的申请数和被通报机构出具的证书数(截至2022年10月)
二、过渡期延长法案
|
器械分类
|
修正法案要求
|
|
Ⅲ 类器械
IIb 类植入器械 (除缝合线钉 (吻合器) 、牙科填充物牙套、牙冠、螺钉、楔子、板、线、针、夹子和连接器)
|
延长至2027年12月31日
|
|
除上述外的所有 Ilb类器械 lla器械以及 I类灭菌或带测量功能的器械
|
延长至2028年12月31日
|
|
在MDD时期不需要公告机构审核,但在MDR时期需要公告机构审核的器械
|
延长至2028年12月31日
|
三、过渡期延长的适用条件
• 医疗器械必须继续遵守指令90/385/EEC或指令93/42/EEC;
• 医疗器械的设计和预期用途没有发生重大变更;
• 医疗器械不会对患者、用户或其他人的健康或安全,或对保护公众健康的其他方面造成不可接受的风险;
• 2024年5月26日之前,制造商必须根据MDR第10(9)条建立质量管理体系(QMS);
• 2024年5月26日之前,制造商或其授权代表需根据MDR附件VII第4.3节提出正式申请,就指令证书或符合性声明涵盖的“遗留设备”,或就MDR下用于替代该设备的设备,进行符合性评估。并且,制造商需和公告机构根据本法规附件VII第4.3节签署书面协议,不迟于2024年9月26日。
原文链接:https://health.ec.europa.eu/system/files/2023-01/mdr_proposal.pdf
中检华通医疗器械质量服务
中检华通威是中国中检下属专业医疗器械检验机构,是集医疗器械产品测试、认证、技术法规咨询、临床验证为一体的“一站式”医疗器械产品质量服务平台。也是唯一一家专门从事检验认证的中央企业。
一、产品检验/检测
电气安全、电磁兼容、产品性能、理化、生物相容性检测、生物安全评价、清洗消毒验证、内毒素、产品有效期验证、临床前功能评价、SAR、无线共存等测试能力。
二、国内注册/全球市场准入
中国NMPA(CFDA)
美国FDA
欧盟CE
澳大利亚TGA
加拿大CMDCAS
英国UKCA
巴西ANVISA (INMETRO,ANATEL )
三、咨询服务
前期产品研发法规辅导
产品技术要求评审服务
临床评价
辅厂建设
质量管理体系建设
转载注明:https://www.szhtw.com.cn/





